Прогресуюча м'язова дистрофія Ерба-Рота - аутосомно-рецесивна спадкова міодистрофія, що відрізняється поліморфізмом клінічних проявів і варіативністю швидкості прогресування симптомів. Може носити спадний характер, тобто починатися зі слабкості в проксимальних відділах рук, але частіше має стандартний висхідний тип поширення м'язових змін. Діагностика здійснюється методами неврологічного огляду, електрофізіологічного тестування, генеалогічного дослідження та генетичного аналізу, за показаннями проводиться гістологічний аналіз біоптату м'язової тканини. Лікування тільки симптоматичне, що дозволяє лише продовжити рухову активність пацієнтів. Підсумок захворювання - повна знедоленість.
Загальна інформація
Прогресуюча м'язова дистрофія Ерба-Рота - найбільш поліморфний варіант спадкової міодистрофії. Від інших видів прогресуючої м'язової дистрофії варіант Ерба-Рота відрізняється варіабельністю як часу дебюту захворювання, так його клінічної картини і течії. Захворювання описано в 1882 р. німецьким неврологом Вільгельмом Ербом. Одночасно в Росії вивченням цієї патології займався В.К. Рот, для позначення міодистрофії він ввів термін «м'язова сухотка». У сучасній світовій і вітчизняній неврології на честь цих дослідників вживається назва «прогресуюча м'язова дистрофія Ерба-Рота». Частота зустрічності захворювання знаходиться в межах від 1,5 до 2,5 випадків на 100 тис. населення.
Прогресуюча м'язова дистрофія Ерба-Рота
Причини міодистрофії Ерба-Рота
Субстратом дистрофії Ерба-Рота є патологічні метаболічні та структурні зміни в м'язовій тканині (міопатія). Вони виникають в результаті генетичних мутацій, що призводять до нестачі або повного припинення синтезу білків, які є необхідними структурними компонентами міоцитів. На сьогоднішній день генетиці відомо не менше 9 хромосомних локусів, аберації в яких призводять до розвитку міодистрофії Ерба-Рота. Найчастіше спостерігаються мутації в локусах 15q15-q21.1, 13q.
Близько 30% генних аномалій виникають de novo, інші носять сімейний характер. Успадковується м'язова дистрофія Ерба-Рота аутосомно-рецесивно. Хворіють як хлопчики, так і дівчатка. Патологія проявляється, якщо дитина отримує аномальний ген від кожного з батьків. У випадку, коли обидва батьки виступають носіями аберрантного гена, але самі не хворіють, ймовірність розвитку міодистрофії у дитини становить 25%.
Симптоми міодистрофії Ерба-Рота
Прогресуюча м'язова дистрофія Ерба-Рота маніфестує в середньому у віці 13-16 років. Однак відомі окремі випадки дебюту хвороби в ранньому дитячому віці і у віці після 20 років. М'язова слабкість і атрофії виникають в першу чергу в мускулатурі тазового поясу і проксимальних відділів ніг. Відзначаються ускладнення ходьби сходами, підйому з положення присів навпочіпки. Типовий симптом Говерса - якщо пацієнт сидів на підлозі, то для того, щоб піднятися, він використовує власне тіло як опору.
З часом дистрофічні зміни поширюються на м'язові групи тулуба і рук. Такий тип поширення міодистрофії носить назву висхідний. Він найбільш типовий для більшості спадкових м'язових дистрофій. Однак у деяких випадках дистрофії Ерба-Рота спостерігається спадний тип поширення патологічного процесу, коли м'язова слабкість виникає спочатку в руках, потім у тазовому поясі, а через кілька років у м'язах ніг.
Тотальна гіпотрофія м'язів тулуба призводить до того, що у пацієнтів починають виступати лопатки (т. зв. «криловидні лопатки»), талія ставати дуже тонкою (т. зв. «осина талія»), посилюється поперековий лордоз, живіт випинається вперед. Характерний симптом вільних надплечий - при спробі підняти хворого вгору, утримуючи його за підмишки, плечі пацієнта вільно рухаються вгору і голова нібито «провалюється» між ними. Ураження лицьових м'язів тягне за собою гіпомімію (т. зв. «обличчя сфінкса»), неповне закриття повік, вивертання і втовщення доль.
Міодистрофія Ерба-Рота супроводжується раннім згасанням колінних рефлексів, а також сухожильних рефлексів з плечового біцепса і трицепсу. Чутлива сфера не порушена. Псевдогіпертрофії не настільки характерні, як при м'язовій дистрофії Беккера. Можуть спостерігатися ретракції сухожиль і м'язові контрактури, але вони виражені меншою мірою, ніж аналогічні прояви м'язової дистрофії Дрейфуса. З часом атрофія і слабкість дихальних м'язів призводить до появи прогресуючої дихальної недостатності, виникає небезпека розвитку застійної пневмонії. Міодистрофічний процес у гладкій мускулатурі обумовлює зниження кишкової перистальтики з тенденцією до запорів. Ураження серцевого м'яза тягне за собою виникнення кардіоміопатії, порушень ритму, серцевої недостатності.
Діагностика міодистрофії Ерба-Рота
Міодистрофія Ерба-Рота діагностується неврологом і генетиком при зіставленні даних анамнезу (вік дебюту захворювання, послідовність розвитку симптомів), неврологічного статусу пацієнта, ЕФІ нервово-м'язової системи, генеалогічних даних, результатів аналізу ДНК і мікроскопічного дослідження м'язової тканини. Диференціювати міодистрофію Ерба-Рота доводиться від інших форм цього захворювання (прогресуючої дистрофії Дюшенна, міодистрофії Дрейфуса і Беккера), дерматоміозиту, поліміозиту, бокового аміотрофічного склерозу, токсичної міопатії та ін.
При неврологічному огляді звертає на себе увагу зниження м'язової сили в мускулатурі проксимальних відділів ніг і рук, гіпотонія і гіпотрофія зазначених м'язів, гіпорефлексія або повне випадання ліктьових і колінних рефлексів, збереження всіх видів чутливості. ЕМГ і ЕНГ свідчать про первинне ураження м'язової тканини при збереженні проведення імпульсів по нервових стовбурах.
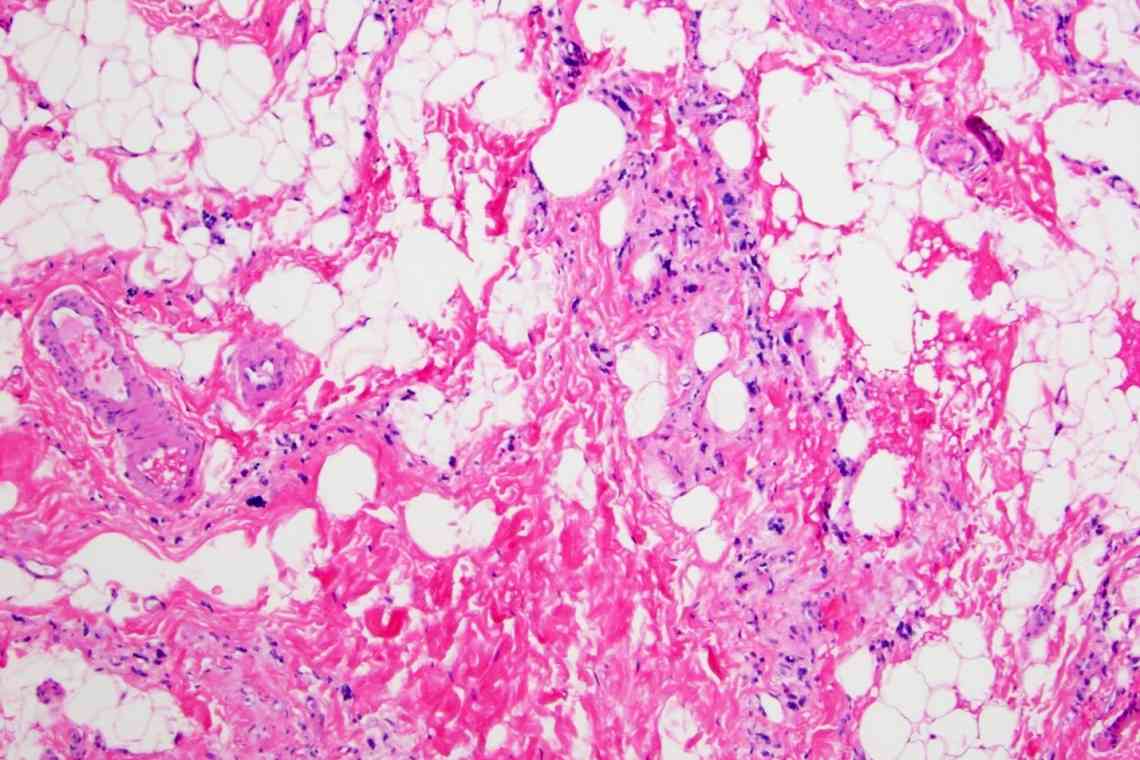
Генеалогічне дослідження підтверджує аутосомно-рецесивний характер спадкування. Дослідження ДНК може виявити наявність генних мутацій. Однак негативний результат дослідження не спростовує діагноз, оскільки не всяка мутація може бути виявлена. Негативний аналіз ДНК є показом до біопсії м'язів. У біоптаті виявляються різні за товщиною м'язові волокна, зменшена кількість м'язових ядер, некротичні і склеротичні зміни.
Різке підвищення рівня креатинфосфокінази характерне для початкового періоду дистрофії Ерба-Рота, потім відбувається поступове зниження цього показника аж до нормальних цифр. Оглядова рентгенографія грудної клітини дозволяє виявити розширення меж серця, наявність запальних змін легеневої тканини. ЕКГ часто визначає аритмію і порушення провідності. За допомогою УЗД серця можна діагностувати кардіоміопатію. Для оцінки ступеня серцевих порушень потрібна консультація кардіолога, при підозрі на пневмонію - консультація пульмонолога.
Лікування міодистрофії Ерба-Рота
Етиопатогенетична терапія поки не розроблена. Симптоматичне лікування спрямоване на якомога більш тривале збереження рухової здатності пацієнта. З цією метою застосовують медикаментозні курси, що включають АТФ, вітаміни Е і групи В, тіоктову кислоту та ін. Заняття лікувальною фізкультурою повинні проводитися щодня і включати вправи на всі групи м'язів. Регулярно призначаються курси масажу та фізіопроцедури.
При ураженні серцевого м'яза рекомендований інозин, серцеві глікозиди, антиаритміки. При розвитку контрактур може знадобитися ортопедичне лікування. Виражене зниження життєвої ємності легенів через атрофію дихальних м'язів служить показанням до ІТЛ.
Прогноз і профілактика
М'язова дистрофія Ерба-Рота може мати різну тяжкість і швидкість прогресування, що буває виражено навіть у межах однієї сім'ї. Описані важкі дюшенноподібні варіанти захворювання з раннім летальним результатом від дихальної недостатності, інфекційних уражень легенів або серцевої недостатності. У відносно легких варіантах міодистрофія може протікати без ураження серцевого м'яза, знедоленість хворих настає лише до 50-річного віку. Профілактикою є своєчасне генетичне консультування сімейних пар, які планують зачаття дитини; виключення близькоспоріднених шлюбів, в яких обидва дружини можуть стати носіями патологічного гена.