Хвороба Гоше - це генетичне захворювання, що характеризується порушенням ліпідного обміну, недостатністю лізосомальних ферментів, накопиченням гліколіпідів у клітинних структурах. Симптоми визначаються типом патології. Загальними ознаками є збільшення печінки, селезінки, зниження згортання крові. При I типі виявляються порушення з боку кісткової системи: остеопороз, часті переломи, інфекції кісток. При II і III типі домінує неврологічна симптоматика: судоми, параліч, косоглазіє, затримка розумового розвитку. Діагностика заснована на біохімічному аналізі дефіцитарного ферменту. Лікування включає ферментозамісну, субстратредуційну та симптоматичну терапію.
Загальна інформація
Захворювання отримало свою назву на прізвище французького лікаря Філіпа Гоше. У 1882 році він описав симптоми і патанатомічні особливості будови селезінки пацієнтки, яка померла від сепсису. Через кілька десятиліть при аналогічному клінічному випадку Гоше визначив накопичення в селезінці глюкоцереброзиду і недостатність ферменту глюкоцереброзідази. Хвороба Гоше (сфінголіпідоз, глюкозилцерамідний ліпідоз) належить до групи лізосомальних хвороб накопичення - спадкових патологій, при яких змінені функції клітинних органелл лізосом. Частота захворювання становить від 1:40 тис. до 1:70 тис. Поширеність найбільш велика в спільнотах, де допустимі шлюби між близькими родичами, наприклад, у євреїв ашкеназі. Носійство мутаційного гена визначається приблизно в 1 людини з 400.
Хвороба Гоше
Причини
Глюкозілцерамідний сфінголіпідоз є найбільш частою формою спадкових ферментопатій. Причиною його розвитку вважається дефект гена GBA, який кодує фермент лізосом бета-глюкозидазу (глюкоцереброзідазу), відповідальну за розщеплення ліпідів. Спадкування хвороби відбувається аутосомно-рецесивним способом, для формування ферментопатії необхідна присутність пари змінених генів: один - від матері, інший - від батька. У подружній парі, де обидва батьки - носії мутації, ймовірність народження хворої дитини становить 25%. Ризик передачі одного дефектного гена, тобто ризик носійства без розвитку хвороби в таких сім'ях дорівнює 50%. При наявності в генотипі двох мутантних алелей функція глюкоцереброзідази знижується на 15-30% від нормального рівня.
Патогенез
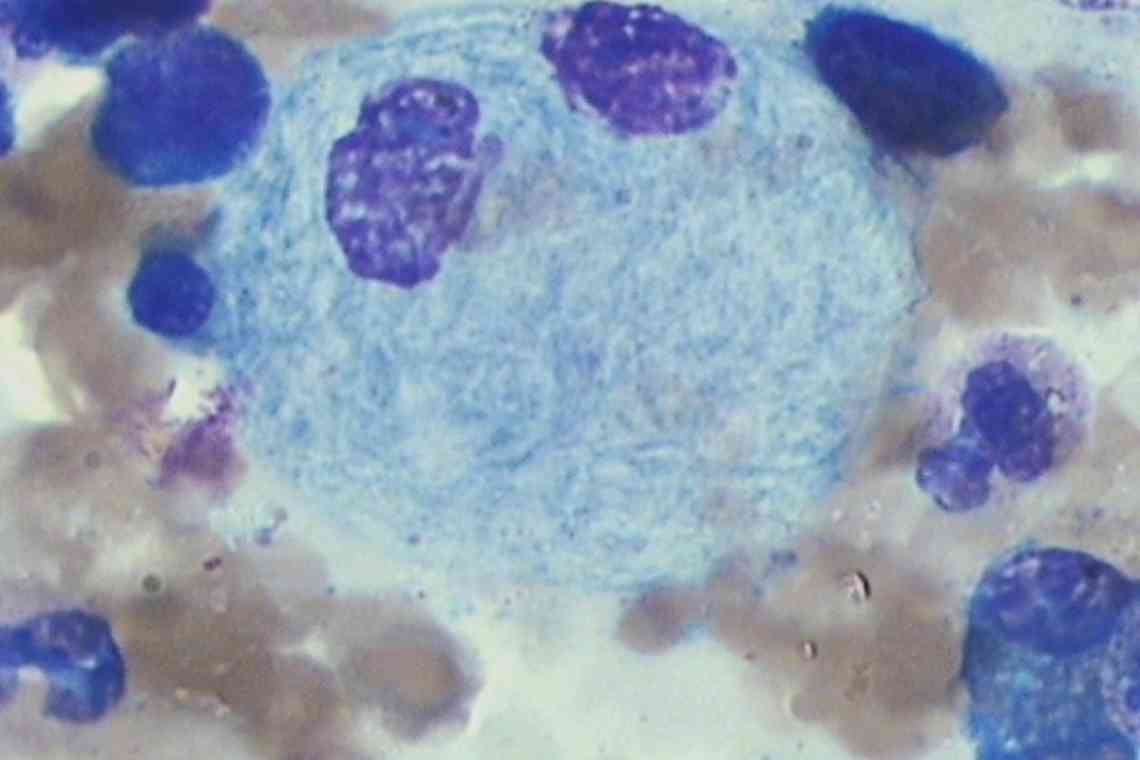
Патогенетичною основою хвороби є зниження каталітичної активності бета-глюкозидази. У результаті порушується процес розщеплення глікосфінголіпідів (складних сполук ліпідів і вуглеводів) до глюкози і цераміду. Аномально прогресивне накопичення макромолекул відбувається в клітинах, які характеризуються підвищеною швидкістю їх оновлення - в макрофагах. Негідролізовані ліпіди концентруються в лізосомах, утворюються особливі клітини накопичення - клітини Гоше. Первинний метаболічний збій провокує вторинні розлади біохімічних процесів і клітинних функцій. Через патологію жирового обміну розвивається синдром активації макрофагів. Стимулюється моноцитопоез, збільшується вміст макрофагів у печінці, селезінці, кістковому мозку. Це стає причиною спленомегалії, гепатомегалії, інфільтрації кісткового мозку. Розлад регуляторної функції макрофагів є провокуючим фактором цитопіння, ураження кісток і суглобів.
Симптоми хвороби Гоше
За віком дебюту і особливостями клінічної картини виділяють три типи хвороби. Перший тип найбільш поширений, має хронічний характер течії. Симптоми частіше проявляються до 30-40 років, рідше хвороба маніфестує в дитячому віці. Збільшення розмірів печінки і селезінки починається відразу після народження, але клінічно проявляється пізніше. Першими ознаками патології стають анемія, підвищена кровоточивість. Гноблення кроветворіння супроводжується зниженням рівня гемоглобіну і тромбоцитів. Зміни з боку опорно-рухового апарату представлені болями в кістках і суглобах, частими переломами, деформаціями (як правило, змінюється стегнова кістка). У дорослих помітна гіперпігментація на обличчі і ногах: шкіра темніє, набуває відтінку від жовтуватого до жовто-коричневого. Може поява плоских червоних плям з типовою локалізацією в області навколо очей. Зростання пацієнтів нижче середнього.
Другий тип хвороби (гострий інфантильний або гострий нейропатичний) зустрічається дуже рідко, розвивається в проміжку від народження до півтора років, найчастіше симптоми дебютують у перші три місяці життя. Характеризується стрімким перебігом, поганим відгуком на лікування. На перший план виходять неврологічні розлади, спровоковані скупченням клітин Гоше в центральній нервовій системі. Діти слабо кричать, мляво смокчуть. Порушено ковтальний рефлекс, нерідко відзначаються збої циклу дихання. Спостерігається помітна затримка психічного та фізичного розвитку. На початковій стадії захворювання м'язовий тонус знижено, через 9-12 місяців після дебюту виникає гіпертонус, особливо в м'язах шиї і кінцівках. Розвиваються судоми, косооке, спастичний параліч. Печінка і селезінка збільшені. Діти часто хворіють на важку пневмонію.
Третій тип - ювенільний або підострий нейропатичний. Перші ознаки - збільшення селезінки і печінки - виникають у 2-3 роки. Повна симптоматика розгортається в період з 6 до 15 років. Клінічні прояви ураження ЦНС включають гіпертонус м'язів, параліч спастичного типу, косоокість, мимовільні спазми, судоми, утруднений цикл дихання з трудністю вдиху, проблеми при ковтанні. Є розлади психічного розвитку: зниження інтелектуальних функцій, несформованість мови і письма, емоційна нестійкість, психози. Діти відстають у статевому розвитку. Перебіг хвороби неухильно прогресує.
Ускладнення
Найбільш важкі ускладнення виявляються при другому і третьому типі хвороби. Ураження спинного і головного мозку призводить до порушення дихального циклу, розвиваються раптові зупинки дихання, зростає ризик спазму гортані і смерті від задухи. Знижений рівень тромбоцитів здатний стати причиною великих внутрішніх кровотечей. У хворих з патологією першого типу поширеним ускладненням є руйнування кісток, їх підвищена ламкість та інфекційні ураження. Обмежується рухливість, пацієнти не можуть пересуватися самостійно, потребують стороннього догляду.
Діагностика
Збір анамнезу і фізикальне обстеження виконується лікарем-ендокринологом і неврологом, додатково призначаються консультації генетика, гематолога, офтальмолога, педіатра, психіатра. Анамнестичні дані включають наявність хвороби Гоше у родичів. При огляді виявляються типові ознаки: низький зріст, патології кісток, неврологічні симптоми (косоокість, атаксія, параліч), геморагічний синдром, гіперпігментація шкіри. Іноді підозра на захворювання виникає після випадкового виявлення збільшеної селезінки на знімках УЗД, пригноблення кровотворної системи за даними загального аналізу крові. Для підтвердження діагнозу, виключення інших метаболічних спадкових патологій, остеомієліту, кісткового туберкульозу, вірусного гепатиту та онкологічних уражень крові проводиться специфічна діагностика:
- Клінічне, біохімічне дослідження крові. У більшості хворих визначається тромбоцитопіння, лейкопенія, анемія, яка у дітей зазвичай має залізодефіцитне походження. У результатах біохімічного аналізу виявляється знижена активність глюкоцереброзідази.
- Ферментний аналіз клітин. При хворобі Гоше в зразках сухої крові і у фібробластах шкіри виявляється недостатня активність глюкозидази. Ступінь дефіцитарності ферменту не має прямої кореляції з вираженістю симптомів. Додатковий біохімічний маркер - хітотріозідаза. Цей фермент синтезується активованими макрофагами, характерно підвищення його активності в 6-10 разів.
- Морфологічне вивчення кісткового мозку. Підтверджується наявність специфічних для даного захворювання структур - клітин Гоше. Результат дозволяє виключити гемобластоз і лімфопроліферативне захворювання.
- Дослідження структури кісткової тканини. З метою оцінки тяжкості ураження кістково-суглобової системи виконується денситометрія, рентгенографія та/або МРТ кісток скелета. Можливий дифузний остеопороз, можуть візуалізуватися колби Ерленмейєра, осередки остеолізису, остеосклерозу і остеонекрозу. На ранніх стадіях хвороби відзначається остеопення, інфільтрація кісткового мозку.
- Візуалізуюче дослідження селезінки, печінки. Проводиться УЗД і МРТ внутрішніх органів. За результатами встановлюється наявність або відсутність осередкових уражень, вимірюється обсяг збільшеного органу. Вихідні показники в подальшому дозволяють контролювати ефективність терапії.
- Молекулярно-генетичні дослідження. ДНК-діагностика є необов'язковою процедурою. Підтвердження мутації в гені GBA буває необхідне при неоднозначності біохімічних досліджень, а також в рамках пренатальних і переваг обстежень.
Лікування хвороби Гоше
Спеціалізована допомога хворим з першим і третім типом хвороби спрямована на усунення симптомів і компенсацію первинного генетичного дефекту - збільшення кількості відсутнього ферменту, посилення катаболізму глікосфінголіпідів. При 2 типі патології терапевтичні заходи виявляються недостатньо ефективними, зусилля лікарів зводяться до полегшення клінічних проявів - болів, судоріг, дихальних розладів. Загальна схема включає наступні напрямки:
- Ферментозамісна терапія. Основним методом лікування є довічна ферментна замісна терапія (ФЗТ) із застосуванням рекомбінантної глюкоцереброзідази. Ефективність досить висока - симптоми повністю купуються, якість життя хворих підвищується. ФЗТ доцільна при третьому і першому типі захворювання. Препарати вводяться внутрішньовенно. Часті інфузії іноді стають причиною запальних захворювань вен (флебітів).
- Субстрат-редукуюча терапія. Даний напрямок є новим у лікуванні хвороби Гоше, відносно широко поширений в США і країнах Європи. Націлено на зниження швидкості виробництва субстрату глікосфінголіпідів і прискорення катаболізму накопичуються макромолекул. В якості препаратів виступають специфічні інгібітори глюкозилцерамідсинтази. Метод показаний при захворюванні 1 типу з легкими і помірними симптомами.
- Симптоматична терапія. При явищах остеопорозу призначається комплексна терапія, що включає прийом кальційовмісних препаратів, вітаміну D і дотримання дієти, збагаченої кальцієм. Ці заходи дозволяють уповільнити втрату кісткової маси, підвищити міцність кісток, запобігти переломам. При скелетних ускладненнях застосовуються анальгезуючі засоби (НПВС), антибактеріальна терапія. Симптоми неврологічних порушень купуються протиепілептичними препаратами, ноотропами, міорелаксантами.
Прогноз і профілактика
Сприятливий результат найбільш вірогідний у пацієнтів з 1 типом захворювання - комплексний терапевтичний підхід дозволяє нормалізувати функціональність глюкоцереброзідази, попередити розвиток ускладнень, уникнути інвалідизації. При 3 типі прогноз залежить від характеру перебігу хвороби, індивідуальної реакції організму на лікувальні заходи. 2 тип має вкрай важкі прояви і завершується загибеллю хворого. Профілактика проводиться під час планування вагітності та на її початкових термінах. Медико-генетичне консультування рекомендується сім'ям, які мають близьких родичів з цією патологією. При високому ризику передачі мутації майбутній дитині в першому триместрі виконується дослідження рівня ферменту в амніотичній рідині, вирішується питання про переривання вагітності.