Хвороба Фабрі - спадкове захворювання, при якому дефект в структурі генів обумовлює недостатню активність або відсутність ферменту лід-галактозидази A, накопичення в органах проміжних продуктів ліпідного обміну. Симптоматика включає болі в кінцівках, зменшення потовиділення, депресію, швидку стомлюваність, ниркову і серцеву недостатність, гострі порушення мозкового кровообігу. Для діагностики використовується дослідження активності ферменту і кількості глікосфінголіпідів в крові і тканинах, секвенування генетичного матеріалу. Лікування ґрунтується на ферментозамісній терапії.
Загальна інформація
Хвороба Фабрі отримала свою назву на прізвище німецького лікаря-дерматолога Джона Фабрі, який у 1898 році детально описав симптоми патології у хлопчика 13 років. В цей же час схожий клінічний випадок був виявлений лікарем-дерматологом з Великобританії Вільямом Андерсеном, тому захворювання має іншу поширену назву - хворобу Андерсона-Фабрі. Менш відомі синоніми - церамідтригексозидоз, дифузна універсальна ангіокератома, спадковий дистонічний ліпідоз. Епідеміологія залежить від расової та етнічної приналежності, становить 1 випадок на 40-120 тис. новонароджених. Найбільш висока поширеність визначається в США.
Хвороба Фабрі
Причини
Захворювання обумовлене мутацією гена GLA, який кодує структуру ферменту альфа-галактозидази А, що бере участь у розщепленні глікосфінголіпідів. Ген локалізований на довгому плечі Xq 22.1 хромосоми. Поліпептидна структура ферменту складається з 429 залишків амінокислот, наявність і порядок розташування яких визначаються 1290 парами підстав гена. У результаті наукових досліджень виявлено 599 варіантів мутацій і поліморфізмів гена, які змінюють стабільність і активність галактозидази. Найпоширеніші причинні мутації - міссенс і нонсенс, на їх частку припадає до 93% випадків хвороби.
Механізм спадкування патології визначений як рецесивний X-зчеплений, але продовжує вивчатися. У гемізіготних чоловіків присутня єдина мутантна X-хромосома. Такий набір представляє класичний фенотип захворювання. Хворі чоловіки здатні передати мутацію тільки дочкам, сини залишаються здоровими. Дочки, які отримали змінені гени від батька, є гетерозіготними і, відповідно до закону рецесивного спадкування, повинні залишатися клінічно здоровими - носіями. Ймовірність передачі патологічного алеля від матері дітям обох статей становить 50%.
Під час клінічних досліджень встановлено, що у більшості жінок з рецесивним мутантним геном хвороба проявляється симптомами помірної вираженості, має пізніше початок і повільне прогресування. У частини пацієнток виявляються важкі прояви патології, які потребують невідкладної медичної допомоги. Причини розвитку важких форм захворювання у гетерозіготних жінок залишаються невідомими. Можливе існування феномена інактивації нормальної X-хромосоми.
Патогенез
Патогенетична основа хвороби - дефіцит альфа-галактози А. У нормі цей фермент розщеплює термінальні гліколіпіди а-галактозилу. При ферментній недостатності в клітинних лізосомах накопичуються проміжні продукти обміну - тригексозилцерамід і дигалактозилцерамід. Вміст церамід-тригекосзиду значно збільшується в ендотеліальних і гладкомишкових клітинах судин, в епітеліальних і перітеліальних клітинах багатьох органів. Молекули дигалактозил-цераміду концентруються в нирках, камерах серця, ЦНС.
Таким чином, основою патогенезу є порушення метаболізму мембранних глікосфінголіпідів. Їх депонування посилюється при підвищеному рівні циркулюючих ліпідів, що надходять в мембрани методом активного всмоктування і дифузії. Основні симптоми захворювання обумовлені скупченням продуктів обміну сфінголіпідів в малих і великих судинах, включають біль в руках і ногах, напади лихоманки, пурпурові шкірні висипання. Патологічні процеси розвиваються поступово, поліморфна клінічна картина часто спостерігається у підлітків і дітей, у дорослих більшою мірою вражається один орган.
Класифікація
Оскільки патологія є генетичною, її початок припадає ще на внутрішньоутробний період. Через відносно повільне прогресування симптоматика проявляється набагато пізніше - до 6-12 або навіть до 30 років. З урахуванням часу дебюту захворювання і характеру ураження внутрішніх органів виділяють дві форми хвороби:
- Класична. Починається в дитинстві і ранньому підлітництві. Проявляється поліорганною поразкою.
- Атипова. Симптоми дебютують у зрілому віці. Часто спостерігається ізольоване пошкодження одного органу: нирок, серця, мозку. У представників чоловічої статі ознаки більш виражені.
- Жіноча. Дана форма виділяється в англомовній літературі. Для неї характерна повільно прогресуюча течія зі слабовираженою симптоматикою, відсутність провідного ураженого органу.
Симптоми хвороби Фабрі
Найбільш частий початковий симптом, який виявляється у 70-80% хворих з класичним варіантом патології - акропарестезії (невропатичні болі). Вони локалізуються в дистальних відділах ступнів і долонь, відрізняються високою інтенсивністю і тривалістю, за характером - пекучі, колючі, виснажливі. Бувають хронічними і кризовими. Хронічні болі постійні, але мають середню інтенсивність. Кризи Фабрі тривають кілька годин або діб, являють собою швидко наростаючі больові напади, що супроводжуються гіпертермією.
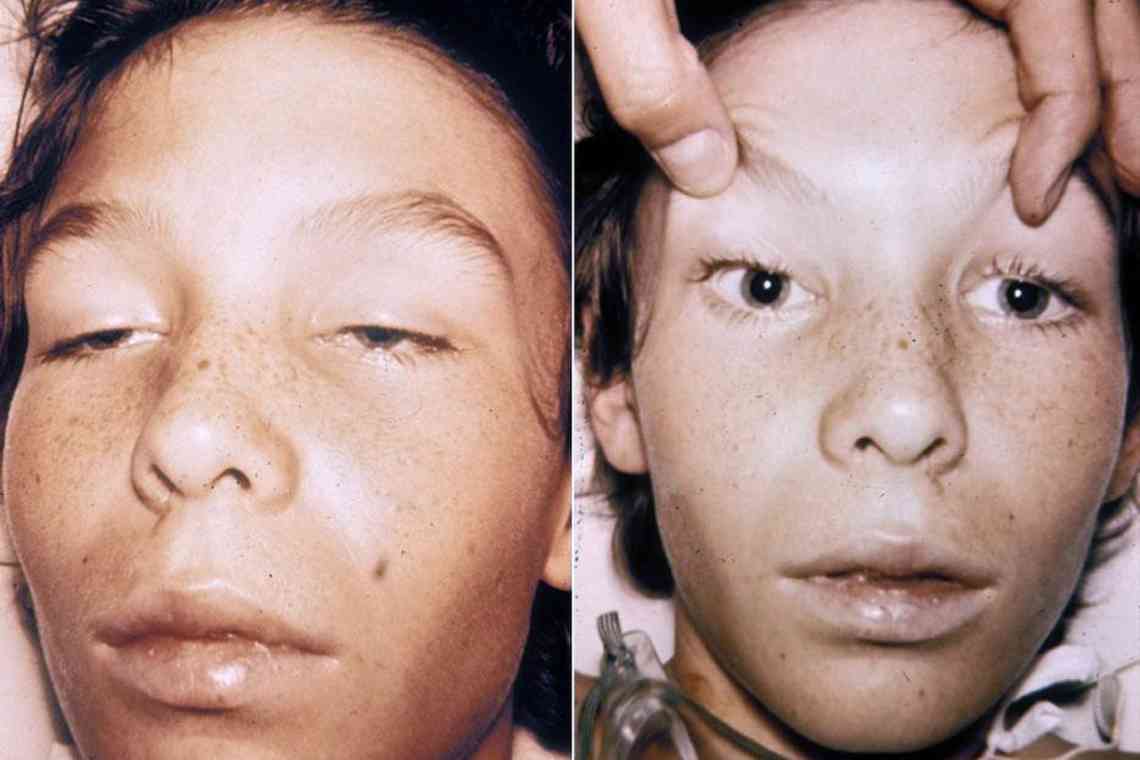
У значній частині хворих спостерігається гіпогідроз і ангідроз - зниження або повна відсутність потовиділення. Пацієнти погано переносять спеку, часто перегріваються (наприклад, при кризових болях). Погіршується працездатність, знижується толерантність до фізичних навантажень. Заняття спортом і фізична праця провокують посилення болів і перегрів тіла. Характерна зовнішня ознака хвороби - ангіокератоми, дрібні безболісні папули червонувато-фіолетового кольору, які розташовуються скупченнями на шкірі. Найчастіше вражається область доль, пальці рук і ніг, геніталії.
Серцево-судинна симптоматика включає аритмії, артеріальну гіпертензію, гіпертрофічну кардіоміопатію. Підлітки страждають від нападів гіпертензії. Дорослі пацієнти відчувають больові відчуття в області грудної клітини, запаморочення, частіше серцебиття і задишку, непритомніють. Пошкодження нирок на початковій стадії протікає безсимптомно. При тривалому перебігу хвороби знижується фільтраційна і концентраційна функція нирок, звернення за лікарською допомогою часто відбувається вже при хронічній нирковій недостатності, що вимагає проведення гемодіалізу.
Неврологічні симптоми більш властиві дорослим. Типові головні болі, запаморочення. Скупчення продуктів метаболізму ліпідів у судинах стає причиною порушення кровообігу в головному мозку. Виникають транзиторні ішемічні атаки та інсульти. Офтальмологічні розлади мають специфічні особливості. Найбільш поширеними є вортексна кератопатія у дітей та помутніння кришталика (двостороння передня та радіальна задня катаракта) у осіб старшого віку. Можливо утворення кон'юнктивальних аневризм, набряк сітківки ока або папілоедему, оптичний неврит з випаданням полів зору, центральними скотомами. Зміни слуху представлені дзвоном у вухах і погіршенням сприйняття звуків. У психоемоційній сфері хворих переважають депресія і тривога.
Ускладнення
Нейропатичні болі при хворобі Фабрі насилу купуються знеболюючими препаратами, тому стають причиною депресії, низької мотивації до навчання, професійної діяльності та іншої соціальної активності. У важких випадках акропарестезії провокуються мінімальним фізичним навантаженням або розумовою перевтомою. У підсумку діти і підлітки переходять на домашнє навчання, що супроводжується звуженням кола друзів, формуванням відчуття ізольованості. Пацієнти перебувають у групі ризику щодо вчинення суїцидальних спроб. Ниркові, серцево-судинні та цереброваскулярні ускладнення можуть призвести до інвалідизації, смерті.
Діагностика
При підозрі на хворобу Фабрі обстеження пацієнта проводиться лікарями декількох спеціальностей: неврологом, кардіологом, дерматологом, генетиком. Діагноз встановлюють на основі типових клінічних проявів, обтяженого сімейного анамнезу, а також за даними лабораторної та інструментальної діагностики. Хворобу Фабрі важливо диференціювати з колагенозами, фіброміалгіями, хворобою Рейно. Специфічні методи дослідження включають:
- Аналіз активності альфа-галактозидази. Досліджують кров (лейкоцити), сухі плями крові, матеріал біопсії нирок, культура шкірних фібробластів. У чоловіків активність ферменту знижена. У жінок можуть визначатися показники, відповідні нормі, нижній межі норми або незначно знижені.
- Кількісний аналіз сфінголіпідів. Виконується дослідження кількості глоботріазилсфінгозину в плазмі крові, сухих плямах крові. Тест широко застосовується при скринінгових обстеженнях. Високі показники з великою ймовірністю вказують на наявність захворювання. Результати використовуються не тільки для підтвердження діагнозу, а й для контролю ефективності лікування.
- Секвенування ДНК. У хворих чоловіків мутації діагностуються в гемізиготному наборі, у хворих жінок або жінок-носіїв - у гетерозіготному. Досліджується ген GLA. Виявлення в його структурі мутаційних змін є найточнішим способом діагностики хвороби Фабрі, особливо щодо жінок.
- МРТ головного мозку. На знімках T2 часто присутній високоінтенсивний сигнал у білій речовині теменної і фронтальної кори. На Т1-зваженому зображенні виявляється гіперінтенсивний сигнал у сірій речовині глибоких структур. Хвороба характеризується ізольованим ураженням заднього бугорка таламусу. Крім того, виявляються судинні мальформації.
- ЕКГ, Ехо-КГ, МРТ серця. Типова гіпертрофія лівого шлуночка, на ранньому етапі захворювання - вкорочення інтервалу P-R, на пізньому етапі - передсердно-шлункова блокада. На МРТ візуалізується пізніше потрапляння контрасту на внутрішню поверхню лівого шлуночка.
Лікування хвороби Фабрі
Єдиним ефективним методом боротьби із захворюванням є ферментозамісна терапія. З початку 2000-х років лікування проводиться рекомбінантними препаратами лід-галактози A. Своєчасна медикаментозна терапія дозволяє знизити вираженість невропатичних болів, зменшити вираженість гіпертрофії лівого серцевого шлуночка, відновити функціональність нирок. Для якнайшвидшого поліпшення самопочуття хворих застосовуються симптоматичні засоби. Парестезії та болі купуються антиконвульсантами, місцевими засобами з лідокаїном.
При недостатності функцій нирок та артеріальної гіпертонії призначаються інгібітори АПФ, блокатори АТ1-рецепторів, гемодіаліз. Для профілактики тромбозів та інсультів використовуються антиагреганти, при тахікардії - антиаритмічні препарати. Методи лікування продовжують розроблятися. В даний час потенційно успішним вважається напрямок генної інженерії. Передбачається, що незабаром стане можливим впровадження в клітини людського організму структурно правильного гена, що задає виробництво альфа суб'єдиниці галактозидази A.
Прогноз і профілактика
Своєчасний початок терапії забезпечує сприятливий перебіг хвороби: симптоми купуються повністю або залишаються слабковираженими, якість життя пацієнтів підвищується, жінки стають здатними зачати і виносити дитину. Без лікування середня тривалість життя чоловіків становить 40-60 років, жінок - 40-70 років. Профілактичні заходи полягають у виявленні носійства мутації, медико-генетичному консультуванні пар, в яких є партнер з підтвердженим діагнозом або обтяженим сімейним анамнезом. При настанні вагітності таким парам необхідно проведення перевагоплантаційної та пренатальної діагностики.