Лейкодистрофія - нейродегенеративне захворювання, обумовлене спадковим порушенням обміну речовин з накопиченням в головному і спинному мозку метаболітів, які провокують руйнування мієліну. Маніфестує в основному в дитячому віці затримкою психомоторного розвитку, руховими розладами, ураженням зорових і слухових нервів, гідроцефалією, епілептичними нападами. Діагностується лейкодистрофія за даними неврологічного статусу, анамнезу, генетичних досліджень, МРТ або КТ картини головного мозку, біохімічних аналізів. Лікування симптоматичне. При ранньому виявленні та повільному прогресуванні можлива трансплантація пуповинної крові або кісткового мозку.
- Загальна інформація
- Причини виникнення лейкодистрофії
- Симптоми лейкодистрофії
- Види лейкодистрофії
- Метахроматична лейкодистрофія залежно від маніфестації має 4 варіанти. Вроджений варіант дебютує в перші 1-3 міс. життя затримкою розвитку і судомним синдромом; діти не досягають віку 1 року. Пізньодетський варіант метахроматичної лейкодистрофії починається в період від 1 до 3 років з м'язової гіпотонії і слабкості, атаксії, затримки психічного розвитку (ЗПР). Потім формується спастична тетраплегія, афазія, псевдобульбарний синдром. У рідкісних випадках пацієнти доживають до 10-річного віку. Ювенільний варіант маніфестує в 4-6 років і триває в середньому 7 років. Дорослий варіант дебютує в третій декаді життя, іноді пізніше, тривалість життя пацієнтів від початку клініки варіює в межах 10-20 років.
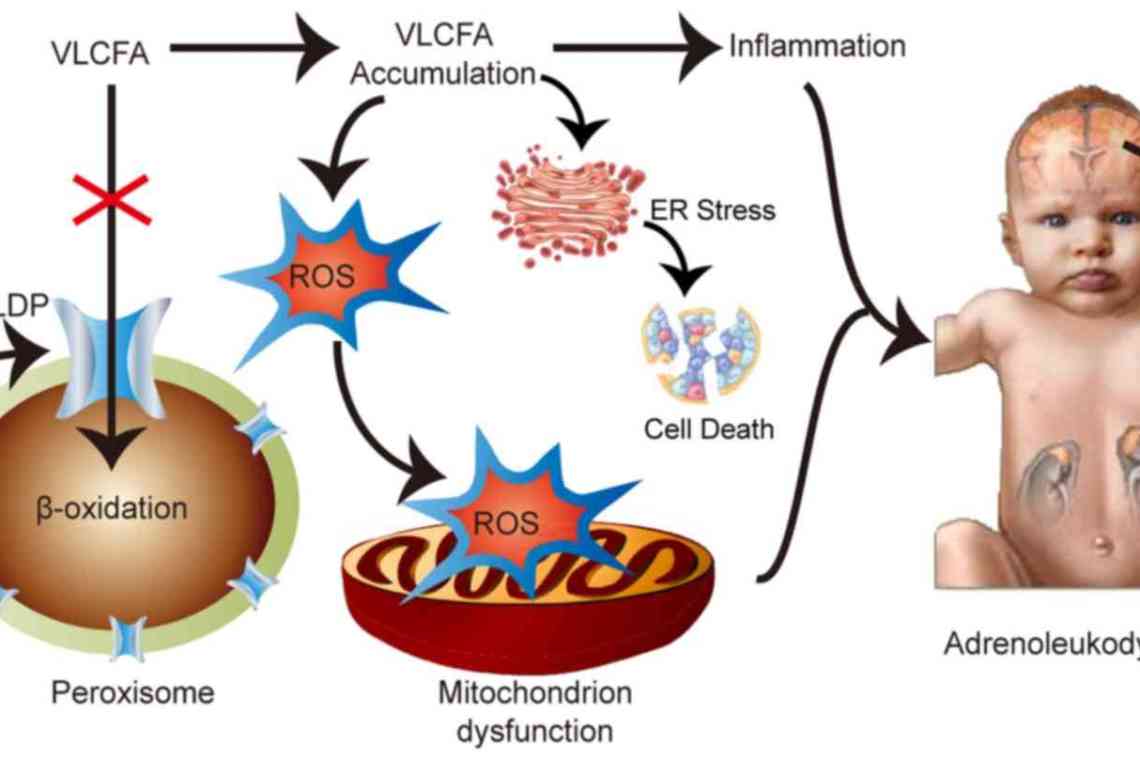
- Суданофільна лейкодистрофія успадковується зчеплено з Х-хромосомою і має кілька різновидів. Лейкодистрофія Пеліцеуса-Мерцбахера може стартувати на 1-му році життя або в 3-4 роки. Першою ознакою є великорозмашистий ністагм, пізніше виникає ЗПР, мозкова атаксія, гіперкінези, парези. Найбільше прогресування відбувається у віці до 10 років, потім захворювання приймає уповільнений перебіг з тривалими ремісіями. Пацієнти можуть жити до зрілого віку. Адренолійкодистрофія - варіант, при якому лейкодистрофія поєднується з наднирковою недостатністю. Характеризується прогресуючою течією з летальним результатом через 6-8 років від початку клініки.
- Глобоїдно-клітинна лейкодистрофія (хвороба Краббе) - ліпоїдоз з накопиченням в осередках демієлінізації галактоцереброзиду і утворенням великих округлих глобоїдних клітин. Ранньодетський варіант розвивається в першому півріччі життя з гіпервозбудності і періодичної гіпертермії, затримується психомоторний розвиток, наростає тонус м'язів, потім розвивається спастичний тетрапарез, олігофренія, епісіндром, можливий опістотонус. У річному віці настає летальний результат. Пізньодетський варіант більш рідкісний, маніфестує погіршенням зору.
- Спонгіозна дегенерація Ван-Богарта-Бертрана характеризується епісіндромом, гіперсомнією, вираженою гідроцефалією зі збільшенням розмірів голови, що викликає амавроз атрофією зорових нервів. Різка внутрішньочерепна гіпертензія призводить до розбіжності черепних швів, реєстрованого при рентгенографії черепа. Пацієнти з цією формою лейкодистрофії гинуть до 3-річного віку.
- Хвороба Александера (лейкодистрофія з волокнистою формацією) обумовлена мутацією гена, відповідального за синтез GFAP білка. У результаті відбувається накопичення в клітинах глії аномального GFAP білка, що містить волокна Розенталя. Неонатальний варіант має важкий перебіг з летальним результатом до кінця 1-го року. Інфантильний варіант зустрічається приблизно в половині випадків, проявляється в перші 1-2 роки життя ЗПР, потім приєднуються спастичні парези, атаксія, гідроцефалія. Діти гинуть через кілька років. Ювенільна лейкодистрофія Александера дебютує в період від 4-х до 10-річного віку, протікає з переважно стовбуровою симптоматикою. Тривалість життя коливається в межах 10-30 років. Дорослий варіант відрізняється пізньою маніфестацією і відносно повільною течією в межах 10 і більше років.
- Лейкодистрофія Галлервордена-Шпатца найчастіше стартує в 10-річному віці. Проявляється дисфункцією стріопалідарної системи, потім на тлі гіперкінезів прогресує тетрапарез, розвивається гемералопія і пігментний ретиніт, спостерігається зниження інтелекту, виникають епіприступи.
- Діагностика лейкодистрофії
- Лікування лейкодистрофії
Загальна інформація
Лейкодистрофія отримала свою назву у зв'язку з ураженням білої речовини мозку (з грецької leukos - білий). Розрізняють близько 60 різновидів лейкодистрофії, що визначаються видом генної аномалії і віком маніфестації клінічних проявів. Поряд з окремими запальними ураженнями ЦНС (наприклад, лейкоенцефалітом Шильдера) лейкодистрофія відноситься до синдрому дифузного склерозу мозку. При цьому домінуюче ураження мієліну зближує її з демієлінізуючими захворюваннями (розсіяним склерозом, РЕМ тощо), а окремі форми можна віднести до ліпідозів.
До основних форм лейкодистрофії належать метахроматична, суданофільна, глобоїдно-клітинна, дегенерація Ван-Богарта-Бертрана, хвороба Александера, варіант Галлервордена-Шпатца. Найбільш поширені перші 3 види лейкодистрофії. Їх зустрічність коливається від 0,4 до 1 випадку на 100 тис. новонароджених. Ряд форм лейкодистрофії є настільки рідкісними, що у світовій літературі з неврології описано всього кілька сотень їх клінічних спостережень. Залежно від вікового періоду, в якому дебютує лейкодистрофія, кожна її форма може підрозділюватися на інфантильний, пізній інфантильний, ювенільний і дорослий варіант.
Лейкодистрофія
Причини виникнення лейкодистрофії
У своїй основі кожна лейкодистрофія має генетичну аномалію певного ферменту. Вид аномалії та локалізація генної мутації поки встановлені лише для найбільш зустрічних форм патології. У більшості випадків лейкодистрофія має аутосомно-рецесивний шлях спадкової передачі, проте окремі її форми можуть успадкуватися зчеплено з підлогою. Крім того, не самотні випадки спонтанних мутацій. Генетично детермінований ензимний дефект веде до обмінних порушень (частіше в метаболізмі ліпідів) з відкладенням певного метаболіту в нервових структурах і окремих соматичних органах, в першу чергу в печінці і нирках.
Наслідком метаболічної аномалії є руйнування мієліну оболонок нервових стовбурів і проводять шляхів, загибель нейронів із заміщенням їх розростається гліальною тканиною. Морфологічно лейкодистрофія характеризується дифузними і симетрично розташованими в півкулях головного мозку зонами загибелі мієліну, скупченням продуктів мієлінового розпаду, посиленою проліферацією глії. В окремих нозологічних варіантах лейкодистрофія має специфічну морфологічну картину - метахроматичне або суданофільне фарбування продуктів мієлінового розпаду, скупчення в зонах демієлінізації глобоїдних клітин тощо.
Симптоми лейкодистрофії
У більшості випадків лейкодистрофія дебютує в ранньому дитячому віці. Новонароджені, як правило, виглядають здоровими. Певний період вони нормально розвиваються, а потім поступово виникають різні неврологічні симптоми, що відрізняються неухильним прогресуванням. Швидкість наростання симптомів тим вища, чим раніше маніфестувала лейкодистрофія. Провідними проявами виступають прогресуюча олігофренія, погіршення зору, тугоухість, епісіндром, спастичні парези. Першими симптомами лейкодистрофії можуть бути атаксія, м'язово-тонічні розлади (гіпо- або гіпертонус, м'язові посмикування), екстрапірамідні прояви, зміни поведінки. Потім виникають епіприступи, бульбарні прояви, знижується слух і зір, відзначається інтелектуальне зниження з поступовою втратою раніше набутих навичок. Сенсорні розлади не характерні. На пізніх етапах розвитку хвороби спостерігаються паралічі, виражена олігофренія, грубий розлад ковтання, амавроз, глухота. У термінальній фазі зазвичай відзначається децеребраційна ригідність.
Види лейкодистрофії
Метахроматична лейкодистрофія залежно від маніфестації має 4 варіанти. Вроджений варіант дебютує в перші 1-3 міс. життя затримкою розвитку і судомним синдромом; діти не досягають віку 1 року. Пізньодетський варіант метахроматичної лейкодистрофії починається в період від 1 до 3 років з м'язової гіпотонії і слабкості, атаксії, затримки психічного розвитку (ЗПР). Потім формується спастична тетраплегія, афазія, псевдобульбарний синдром. У рідкісних випадках пацієнти доживають до 10-річного віку. Ювенільний варіант маніфестує в 4-6 років і триває в середньому 7 років. Дорослий варіант дебютує в третій декаді життя, іноді пізніше, тривалість життя пацієнтів від початку клініки варіює в межах 10-20 років.
Суданофільна лейкодистрофія успадковується зчеплено з Х-хромосомою і має кілька різновидів. Лейкодистрофія Пеліцеуса-Мерцбахера може стартувати на 1-му році життя або в 3-4 роки. Першою ознакою є великорозмашистий ністагм, пізніше виникає ЗПР, мозкова атаксія, гіперкінези, парези. Найбільше прогресування відбувається у віці до 10 років, потім захворювання приймає уповільнений перебіг з тривалими ремісіями. Пацієнти можуть жити до зрілого віку. Адренолійкодистрофія - варіант, при якому лейкодистрофія поєднується з наднирковою недостатністю. Характеризується прогресуючою течією з летальним результатом через 6-8 років від початку клініки.
Глобоїдно-клітинна лейкодистрофія (хвороба Краббе) - ліпоїдоз з накопиченням в осередках демієлінізації галактоцереброзиду і утворенням великих округлих глобоїдних клітин. Ранньодетський варіант розвивається в першому півріччі життя з гіпервозбудності і періодичної гіпертермії, затримується психомоторний розвиток, наростає тонус м'язів, потім розвивається спастичний тетрапарез, олігофренія, епісіндром, можливий опістотонус. У річному віці настає летальний результат. Пізньодетський варіант більш рідкісний, маніфестує погіршенням зору.
Спонгіозна дегенерація Ван-Богарта-Бертрана характеризується епісіндромом, гіперсомнією, вираженою гідроцефалією зі збільшенням розмірів голови, що викликає амавроз атрофією зорових нервів. Різка внутрішньочерепна гіпертензія призводить до розбіжності черепних швів, реєстрованого при рентгенографії черепа. Пацієнти з цією формою лейкодистрофії гинуть до 3-річного віку.
Хвороба Александера (лейкодистрофія з волокнистою формацією) обумовлена мутацією гена, відповідального за синтез GFAP білка. У результаті відбувається накопичення в клітинах глії аномального GFAP білка, що містить волокна Розенталя. Неонатальний варіант має важкий перебіг з летальним результатом до кінця 1-го року. Інфантильний варіант зустрічається приблизно в половині випадків, проявляється в перші 1-2 роки життя ЗПР, потім приєднуються спастичні парези, атаксія, гідроцефалія. Діти гинуть через кілька років. Ювенільна лейкодистрофія Александера дебютує в період від 4-х до 10-річного віку, протікає з переважно стовбуровою симптоматикою. Тривалість життя коливається в межах 10-30 років. Дорослий варіант відрізняється пізньою маніфестацією і відносно повільною течією в межах 10 і більше років.
Лейкодистрофія Галлервордена-Шпатца найчастіше стартує в 10-річному віці. Проявляється дисфункцією стріопалідарної системи, потім на тлі гіперкінезів прогресує тетрапарез, розвивається гемералопія і пігментний ретиніт, спостерігається зниження інтелекту, виникають епіприступи.
Діагностика лейкодистрофії
Діагностичний пошук вимагає залучення ряду фахівців: невролога, педіатра, медичного генетика, для діагностики розладів зору і слуху - отоларинголога і офтальмолога. Важливе значення має вивчення анамнезу хвороби (вік і симптоми дебюту, послідовність розвитку клініки) і сімейного анамнезу (наявність лейкодистрофії у родичів). Нейросонографія через нічок і відлуння енцефалографія у пацієнтів більш старшого віку, як правило, виявляє підвищення інтракраніального тиску. Лейкодистрофія супроводжується істотним збільшенням концентрації білка, обумовленим руйнуванням церебральних клітин, що визначається при дослідженні цереброспинальної рідини.
З метою діагностики виду метаболічної аномалії проводиться цілий ряд біохімічних тестів з визначенням рівня ферментів і накопичуваних метаболітів. Осередки демієлінізації добре візуалізуються за допомогою МРТ, можуть бути виявлені і на КТ головного мозку. Зазвичай демієлінізацію видно на МРТ головного мозку ще до клінічної маніфестації лейкодистрофії. Завдяки розвитку генетики, лейкодистрофія має розроблену ДНК-діагностику, а окремі її форми (метахроматична, адренолійкодистрофія, глобоїдно-клітинна) - можливість пренатального діагностування.
Лікування лейкодистрофії
На сьогоднішній день лейкодистрофія не має ефективних способів терапії, що дозволяють купірувати прогресування симптомів. Проводиться симптоматичне лікування - в основному дегідратаційна та антиконвульсантна терапія. Єдиним методом, здатним збільшити тривалість життя пацієнтів з лейкодистрофією і поліпшити якість їх життя, є трансплантація пуповинної крові або пересадка кісткового мозку. Трансплантація призводить до нормалізації метаболізму. Однак цей процес займає тривалий час (від 12 до 24 міс.), протягом якого триває прогресування лейкодистрофії. Тому найчастіше важка інвалідизація або загибель пацієнта настає навіть після успішної трансплантації.
Слід підкреслити, що трансплантація ніяк не впливає на неврологічний дефіцит, що вже розвинувся, вона лише дозволяє призупинити його подальше прогресування. У зв'язку з тим, що ефект такого лікування настає через 1-2 роки, воно доцільне у разі ранньої доклінічної діагностики лейкодистрофії (при відповідній настороженості батьків народженої дитини у зв'язку з наявністю подібної патології в сім'ї) або при повільно прогресуючому варіанті перебігу. Крім того, необхідно враховувати, що трансплантація пов'язана з ризиком ряду серйозних ускладнень, таких як відторгнення, реакція «трансплантат проти господаря», розвиток інфекцій.