Амавроз Лебера - це спадкове захворювання, що характеризується вродженим ураженням світлочутливих клітин сітківки ока і в деяких випадках іншими загальними порушеннями (аномалії нирок, ЦНС). При цій патології в перші місяці життя дитини або відразу після народження з'являється ністагм, ослаблення або відсутність реакції зіниці на світ. Надалі дитина може терти очі (симптом Франческетті), виникає дальнозоркість і світлобоязнь, можлива повна втрата зору. Діагностика ґрунтується на даних огляду пацієнта лікарем-офтальмологом, електроретинографії, дослідження спадкового анамнезу і генетичних аналізів. Специфічне лікування амавроза Лебера на сьогоднішній день не розроблено.
Загальна інформація
Вроджений амавроз Лебера являє собою гетерогенну групу захворювань, причиною яких виступають мутації в 18 генах, що кодують різні білки сітківки, в тому числі опсин. Вперше амавроз був описаний ще в XIX столітті (в 1867 році) Т. Лебером, який вказав основні прояви цього захворювання - маятниковий ністагм, сліпота, поява пігментних плям і включень на очному дні. Середня поширеність захворювання становить 3:100000 населення.
Амавроз Лебера рівною мірою вражає як чоловіків, так і жінок. Захворювання становить приблизно 5% від усіх спадкових ретинопатій. Сучасна генетика розробляє методики лікування цієї патології, є обнадійливі результати генної терапії однієї з форм амавроза Лебера, обумовленої мутацією в гені RPE65. Окремо виділяють атрофію зорових нервів Лебера, яка також характеризується поступовою втратою гостроти зору і згодом повною сліпотою. Однак це захворювання зовсім іншої генетичної природи і обумовлене пошкодженням мітохондріальної ДНК, яка має свій унікальний тип спадкування (по материнській лінії).
Амавроз Лебера
Причини
Основний механізм розладу зору при амаврозі Лебера - порушення метаболізму в паличках і колбочках, яке веде до летальних пошкоджень фоторецепторів і їх руйнування. Однак безпосередня причина таких змін розрізняється залежно від того, мутація якого саме гена викликала захворювання.
Один з найбільш поширених типів амавроза Лебера (тип 2, LCA2) обумовлений наявністю мутантного гена RPE65 на першій хромосомі. Відомо більше 80-ти мутацій цього гена, деякі з яких, крім амавроза Лебера, викликають і певні форми пігментної абіотрофії сітківки. Білок, кодований PRE65, відповідає за метаболізм ретинолу в пігментному епітелії сітчастої оболонки ока, тому при наявності генетичного дефекту цей процес порушується з розвитком побічних метаболічних шляхів. В результаті цього синтез родопсину у фоторецепторах припиняється, що і призводить до характерної клінічної картини захворювання. Мутантні форми гена наслідуються за аутосомно-рецесивним механізмом.
Менш поширена форма амавроза Лебера (тип 14) викликана мутацією гена LRAT на 4-й хромосомі. Він кодує білок лецитин-ретинол-ацилтрансферазу, який розташовується в мікросомах гепатоцитів і виявлений у сітківці ока. Цей фермент бере участь у метаболізмі ретиноїдів і вітаміну А, через наявність мутацій у гені отриманий протеїн не може повноцінно виконувати свої функції, через що розвивається дегенерація фоторецепторів, яка клінічно проявляється амаврозом Лебера або ювенільною пігментною абіотрофією сітківки. Має аутосомно-рецесивний характер спадкування.
Амавроз Лебера тип 8 найбільш часто призводить до вродженої сліпоти, відповідальний за розвиток цієї форми захворювання ген CRB1 розташовується на 1-й хромосомі і має аутосомно-рецесивний характер спадкування. При цьому з'ясовано, що кодований цим геном білок бере безпосередню участь в ембріональному розвитку фоторецепторів і пігментного епітелію сітківки. Більш точних даних по патогенезу даної форми амавроза Лебера на сьогоднішній день не накопичено. Аналогічна ситуація з мутацією гена LCA5, розташованого в 6-й хромосомі і асоційованого з 5-м типом амаврозу. В даний час виявлено тільки білок, кодованим даним геном - леберцилін, але його функції в сітківці незрозумілі.
Також виявлено дві форми амавроза Лебера, які наслідуються за аутосомно-домінантним механізмом - тип 7, обумовлений мутацією гена CRX, і тип 11, асоційований з порушенням гена IMPDH1. Ген CRX кодує білок, який володіє безліччю функцій - контроль розвитку фоторецепторів в ембріональний період, підтримання їх адекватного рівня в дорослому віці, участь в синтезі інших протеїнів сітківки (є фактором транскрипції). Тому залежно від характеру мутації гена CRX клініка амавроза Лебера 7-го типу може бути різноманітною - від вродженої сліпоти до відносно пізнього і уповільненого погіршення зору.
Інозин-5'-монофосфатдегідрогеназа 1, кодований геном IMPDH1, являє собою фермент, що регулює зростання клітин і утворення нуклеїнових кислот, проте це поки не дозволяє прояснити патогенез того, як порушення цього білка призводять до 11-го типу амавроза Лебера.
Класифікація амавроза Лебера
Зараз повністю доведено взаємозв'язок між клінічними проявами і мутаціями певних генів для 16-ти типів амаврозу Лебера. Також є вказівки про відкриття ще двох генів, пошкодження в яких призводять до такого захворювання, але поки в цьому відношенні проводяться додаткові дослідження.
- Тип 1 (LCA1, від англійського leber's congenital amaurosis) - пошкоджений ген GUCY2D на 17-й хромосомі, тип спадкування аутосомно-рецесивний.
- Тип 2 (LCA2) - пошкоджений ген RPE65 на 1-й хромосомі, аутосомно-рецесивне спадкування, є перші позитивні результати з генної терапії цієї форми амавроза Лебера.
- Тип 3 (LCA3) - пошкоджений ген RDH12 на 14-й хромосомі, аутосомно-рецесивне успадкування.
- Тип 4 (LCA4) - пошкоджений ген AIPL1 на 17-й хромосомі, аутосомно-рецесивне успадкування.
- Тип 5 (LCA5) - пошкоджений ген LCA5 на 6-й хромосомі, аутосомно-рецесивне успадкування.
- Тип 6 (LCA6) - пошкоджений ген RPGRIP1 на 14-й хромосомі, аутосомно-рецесивне спадкування.
- Тип 7 (LCA7) - пошкоджений ген CRX на 19-й хромосомі, аутосомно-домінантне спадкування. Характеризується варіабельною клінічною картиною.
- Тип 8 (LCA8) - пошкоджений ген CRB1 на 1-й хромосомі, аутосомно-рецесивне успадкування. Статистично частіше за інші типи призводить до вродженої сліпоти.
- Тип 9 (LCA9) - пошкоджений ген LCA9 на 1-й хромосомі, аутосомно-рецесивне успадкування.
- Тип 10 (LCA10) - пошкоджений ген CEP290 на 12-й хромосомі, аутосомно-рецесивне успадкування.
- Тип 11 (LCA11) - пошкоджений ген IMPDH1 на 7-й хромосомі, аутосомно-домінантне успадкування.
- Тип 12 (LCA12) - пошкоджений ген RD3 на 1-й хромосомі, аутосомно-рецесивне успадкування.
- Тип 13 (LCA13) - пошкоджений ген RDH12 на 14-й хромосомі, аутосомно-рецесивне успадкування.
- Тип 14 (LCA14) - пошкоджений ген LRAT на 4-й хромосомі, аутосомно-рецесивне спадкування.
- Тип 15 (LCA15) - пошкоджений ген TULP1 на 6-й хромосомі, аутосомно-рецесивне успадкування.
- Тип 16 (LCA16) - пошкоджений ген KCNJ13 на 2-й хромосомі, аутосомно-рецесивне успадкування.
Крім того, іноді в клінічній класифікації виділяють не тільки назву пошкодженого гена, але і характер мутації, оскільки це має значний вплив на перебіг амавроза Лебера. Більш того, різні типи мутацій в одному і тому ж гені можуть призводити до абсолютно різних захворювань - наприклад, деякі різновиди делецій в гені CRX можуть призводити не до амаврозу, а до паличко-колбочкової дистрофії. Деякі мутації генів RPE65, LRAT і CRB1 є причиною різних форм пігментної абіотрофії сітківки.
Симптоми амавроза Лебера
Симптоматика амавроза Лебера досить варіабельна і залежить від типу захворювання і характеру мутації гена. У більшості випадків при народженні дитини патологія не визначається - навіть при огляді очного дна зміни спостерігаються лише в декількох відсотках випадків. У міру його зростання батьки можуть помічати, що дитина не затримує погляд на предметах і оточуючих, а в більш старшому віці може болісно реагувати на світло (з'являється фотофобія), часто терти очі і вказувати на них пальцем (симптом Франческетті, окулопальцевий синдром). Виявляється ністагм, який виникає ще в перші 2-3 місяці життя і часто є одним з перших проявів амавроза Лебера, уповільнена реакція зіниці на світло або її повну відсутність.
У ряді випадків спостерігається вроджена сліпота. Якщо ж дитина народилася з відносно збереженою функцією зору, то в перші роки життя, крім зазначених симптомів, у її також розвивається дальнозоркість, косоокість, сильно страждає гострота зору. Зазвичай до 10-ти років більшість хворих з амаврозом Лебера повністю зліпнуть. Надалі у них можуть виникати й інші порушення зорового апарату - кератоконус, катаракта, глаукома. При деяких типах захворювання можуть спостерігатися і супутні порушення - ураження ЦНС, глухота.
Діагностика
У сучасній офтальмології діагностика амавроза Лебера проводиться на підставі огляду очного дна, моніторингу динаміки змін у ньому, даних електроретинографії. Важливу роль відіграє також вивчення спадкового анамнезу, а для деяких типів захворювання - генетичне секвенування послідовності ключових генів.
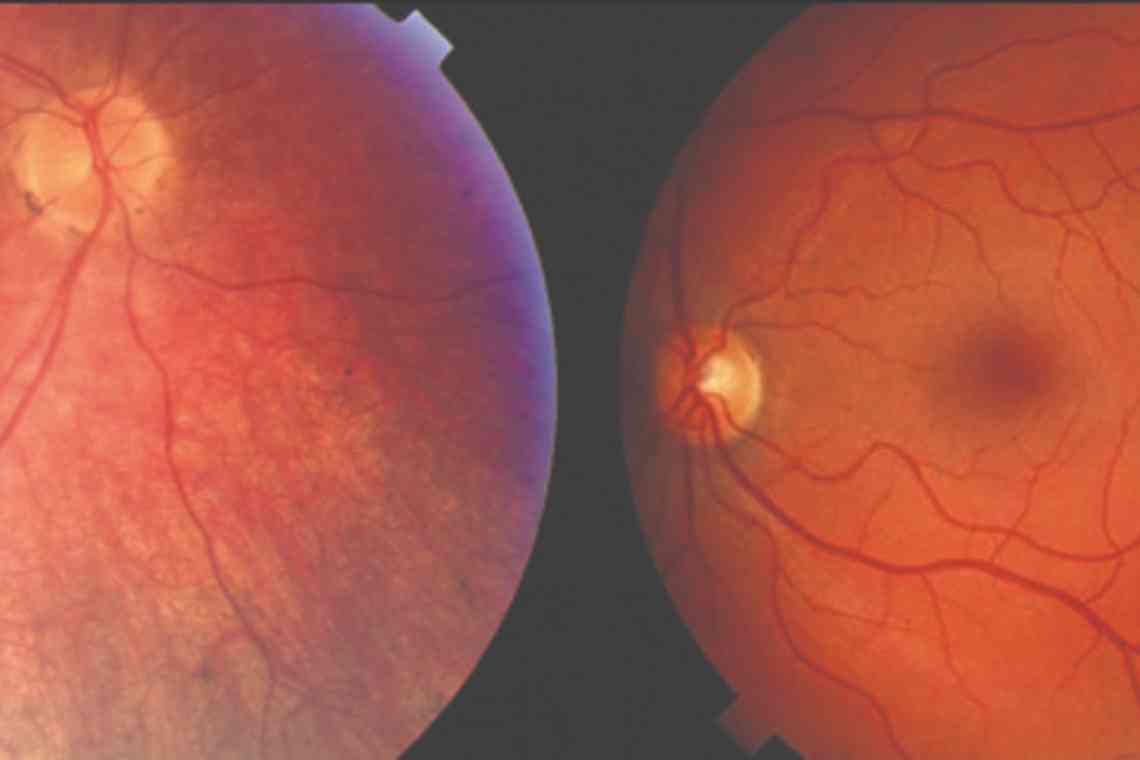
- Офтальмоскопія. При огляді очного дна відносно довгий час (перші кілька років життя) ніяких змін може не реєструватися. Першими, але не специфічними офтальмологічними симптомами амаврозу є ністагм, косоокість, уповільнена або відсутня реакція зіниць на світло. З часом зміни сітківки зводяться до появи пігментних або непигментованих плям різного розміру, звуження артеріол, блідості диска зорового нерва. До 8-10 років практично у всіх хворих спостерігаються кісткові пігментні тільця, розташовані по периферії очного дна.
- Візометрія. Характерною ознакою є більш швидке прогресування змін на сітківці порівняно з функціональними порушеннями зору, які розвиваються відносно повільно. До розвитку сліпоти гострота зору становить 0,1 і менш, часто реєструється дальнозоркість, світлобоязнь.
- Електроретинографія. ЕРГ при амаврозі Лебера, як правило, відображає сильне зниження амплітуди всіх хвиль або їх повну відсутність.
- Генетичні дослідження. Дозволяють виявити пошкоджений ген і тип мутації тільки в 50-60% випадків (частота найбільш поширених пошкоджень генів). Переважна більшість клінік проводять секвенування послідовностей з метою виявлення мутацій тільки щодо генів RPE65, CRX, CRB1, LCA5 і KCNJ13.
Диференціальну діагностику виробляють з різними формами пігментної абіотрофії сітківки (при ній зберігається нормальна або трохи знижена амплітуда хвиль на електроретинограмі) і деякими типами атрофії зорових нервів.
Лікування амавроза Лебера
На сьогоднішній день специфічного лікування будь-якого типу амавроза Лебера не існує. На етапі клінічних випробувань знаходиться генно-інженерне введення гена RPE65 в сітчасту оболонку ока хворих на амавроз 2-го типу, є перші дані про значне поліпшення зору піддослідних хворих. У разі ж інших форм захворювання такого прогресу поки немає. Підтримувальне лікування зводиться до вітамінної терапії, внутрішньоочних ін'єкцій судинних засобів. При дальнозоркості призначається носіння очок.
Прогноз
У плані збереження зору прогноз вкрай несприятливий, практично 95% хворих повністю втрачають здатність бачити до 10-го року життя. Крім того, це спадкове захворювання може ускладнюватися проблемами з ЦНС, нирками, ендокринною системою, що вимагає більш ретельного медичного моніторингу для своєчасного виявлення подібних порушень.